soticlestat
overview
Soticlestat is a potential first-in-class oral medicine that is intended to reduce seizure susceptibility and improve seizure control for people living with Lennox-Gastaut syndrome and Dravet syndrome. The two Phase 3 trials evaluating efficacy and safety of soticlestat have completed and top line data was reported by Takeda on June 17, 2024. Ovid co-developed soticlestat through Phase 2 and then sold its rights to Takeda. Ovid retains financial interests to soticlestat, click here»
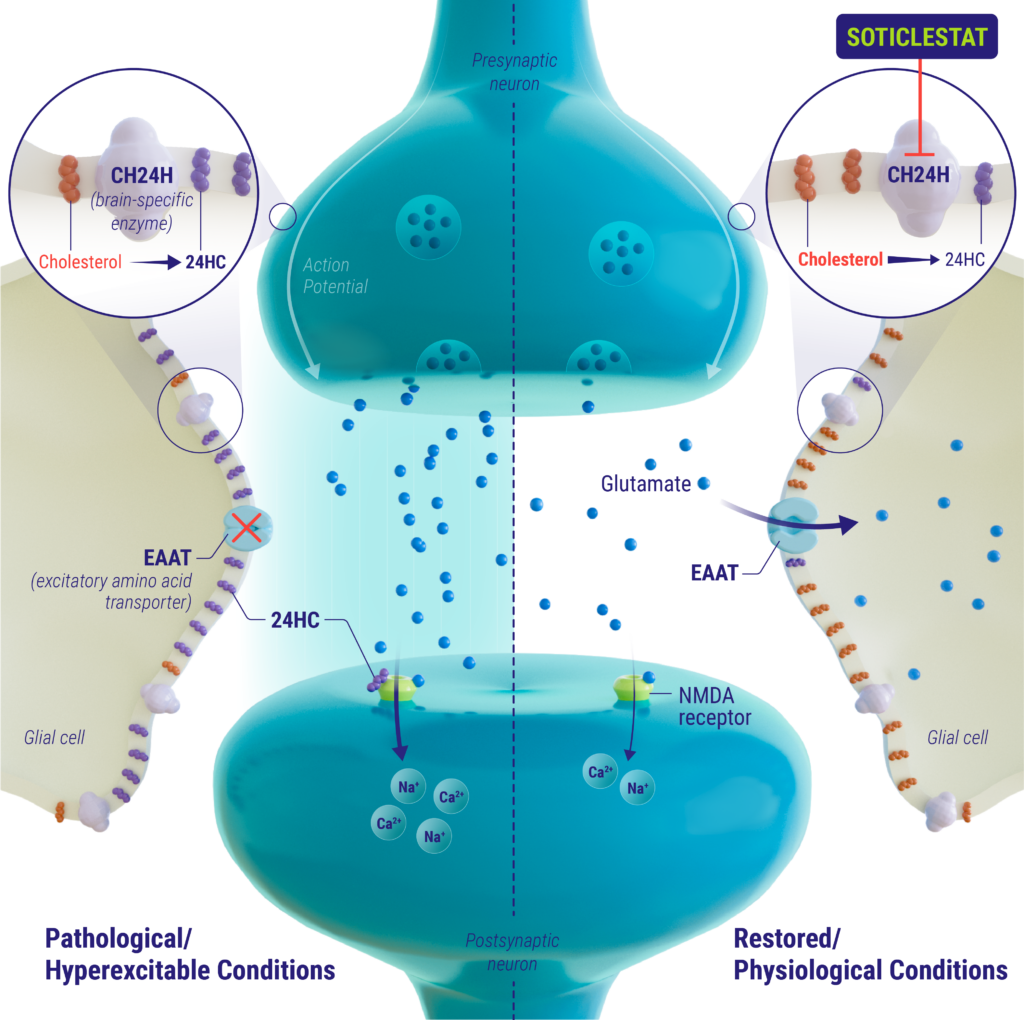
mechanism of action
Soticlestat is a potent, highly selective inhibitor of the brain-specific enzyme cholesterol 24-hydroxylase (CH24H). CH24H is predominantly expressed in the brain where it converts cholesterol into 24S-hydroxycholesterol (24HC) to adjust the homeostatic balance of brain cholesterol. 24HC has multiple functions in the brain, one of which is as a positive allosteric modulator of the NMDA receptor. Glutamate is the main excitatory neurotransmitter in the brain and has been shown to play a role in the initiation and spread of seizure activity. Studies suggest that 24HC is involved in over-activation of the glutamatergic pathway through modulation of the NMDA receptor and disruption of astrocytic glutamate amino acid transporter 2 (EAAT2), resulting in epileptogenesis. Inhibition of CH24H by soticlestat reduces the neuronal levels of 24HC and may improve the excitatory/inhibitory balance in the brain, thereby reducing seizure susceptibility and improving seizure control.
development
Ovid Therapeutics designed and conducted a robust clinical program for soticlestat in individuals with developmental and epileptic encephalopathies (DEE). A Phase 1b/2a study in adults with DEE yielded positive results. Additionally, Ovid completed two Phase 2 studies with soticlestat:
- The Phase 2 ARCADE study in pediatric patients with CDKL5 deficiency disorder or Dup15q syndrome was completed and yielded positive results.
- The Phase 2 ELEKTRA study in pediatric patients with Lennox-Gastaut syndrome or Dravet syndrome was completed and yielded positive results.
Takeda led two Phase 3 studies in children and adults with Dravet syndrome (NCT04940624) and Lennox-Gastaut syndrome (NCT04938427) and the open-label ENDYMION extension study (NCT05163314).
For additional information on soticlestat clinical and non-clinical data, please click here »
Takeda will engage with regulatory authorities to discuss the totality of the data generated by these studies to determine next steps. Takeda will also plan to present results of both Phase 3 studies in DS and LGS at an upcoming scientific congress. See below to learn more about these syndromes:
Dravet syndrome
- Dravet syndrome is most commonly caused by a genetic mutation in the SCN1A gene
- Dravet syndrome is estimated to affect approximately 1 in 15,000 in the US (Wu et al 2015)
- Dravet syndrome is characterized by prolonged focal seizures that can evolve to convulsive tonic-clonic seizures
- Children with Dravet syndrome experience developmental disabilities as seizures increase. Other common symptoms include changes in appetite, difficulty balancing and a crouched gait when walking.
Reference: 1. Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, Kuzniewicz MW. Incidence of Dravet Syndrome in a US Population. Pediatrics. 2015
Lennox-Gastaut syndrome
- Lennox-Gastaut syndrome is estimated to affect approximately 1 in 11,000 individuals in the US (Trevathan, 1997)
- Lennox-Gastaut syndrome is a heterogeneous condition, characterized by several different types of seizures, most commonly atonic (drop), tonic and atypical absence seizures
- Children with Lennox-Gastaut syndrome may also develop cognitive dysfunction, delays in reaching developmental milestones and behavioral problems
Reference: 1. Trevathan E, Murphy CC, Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia. 1997